Roles
One of the benefits of a career in regulatory affairs is the variety of options and flexibility that skilled and experience professionals potentially have open to them. From working as an independent contractor to rising through the ranks in a big pharma company. Here is an illustration of the career progression that a regulatory professional could achieve:
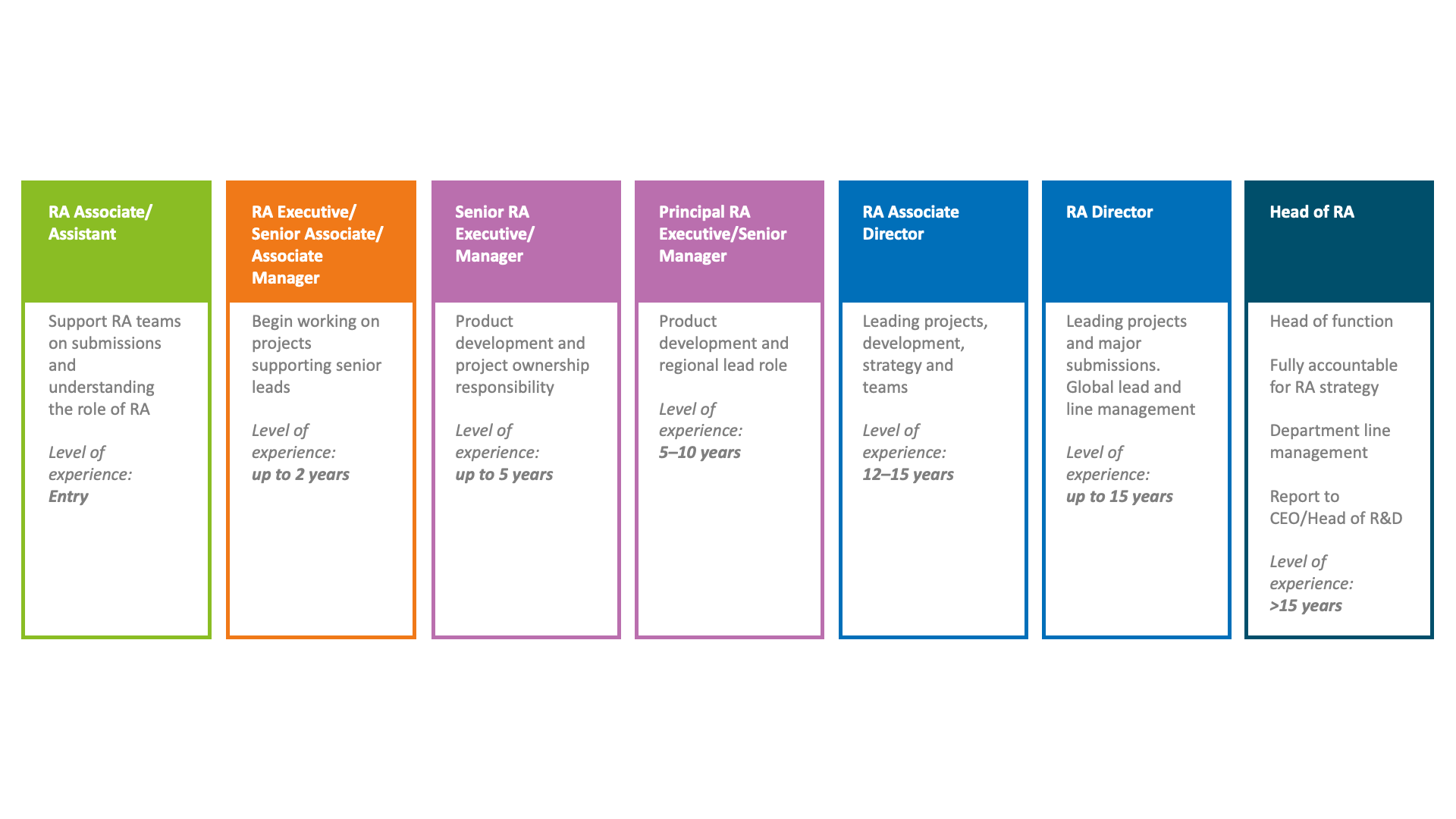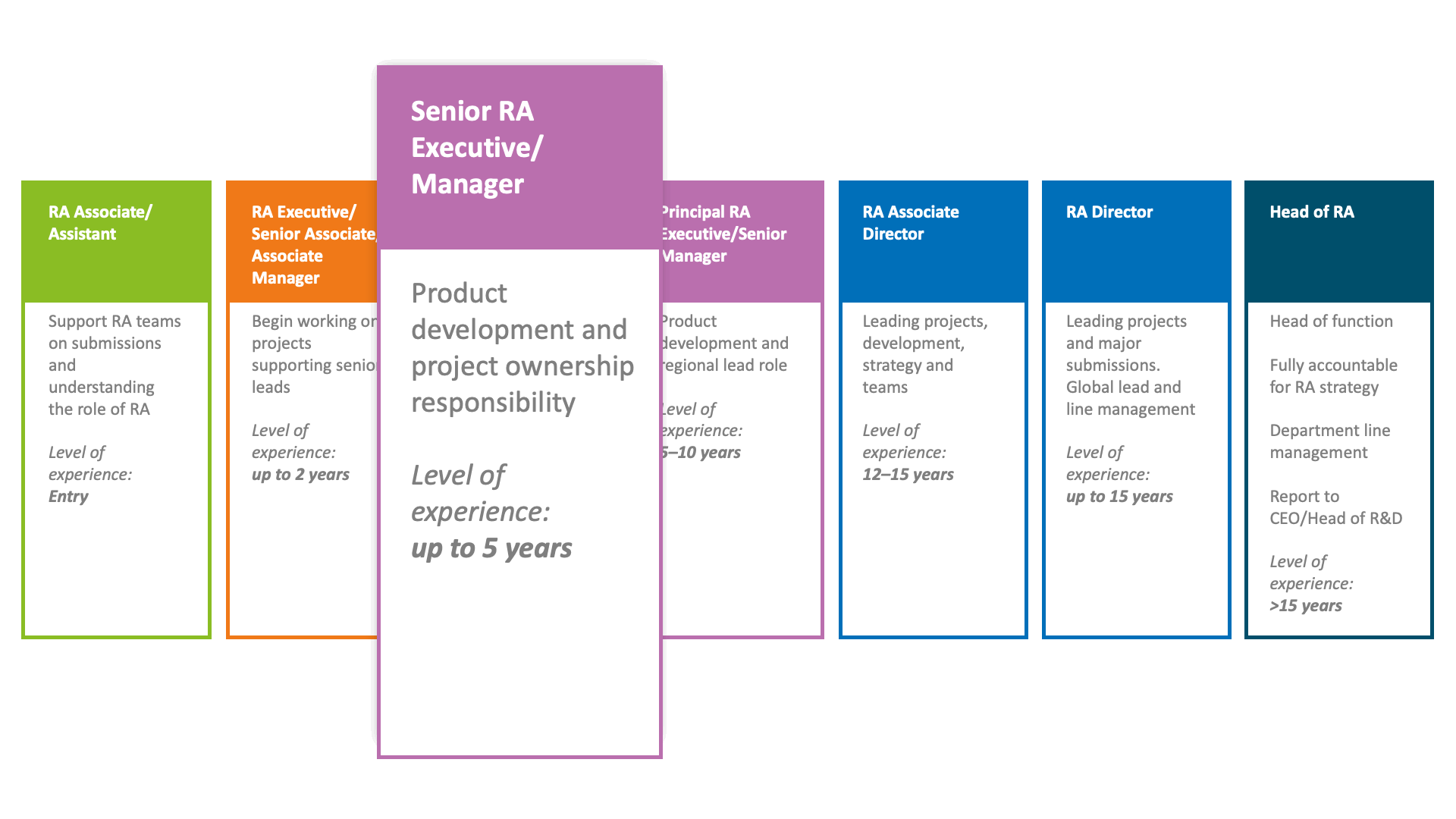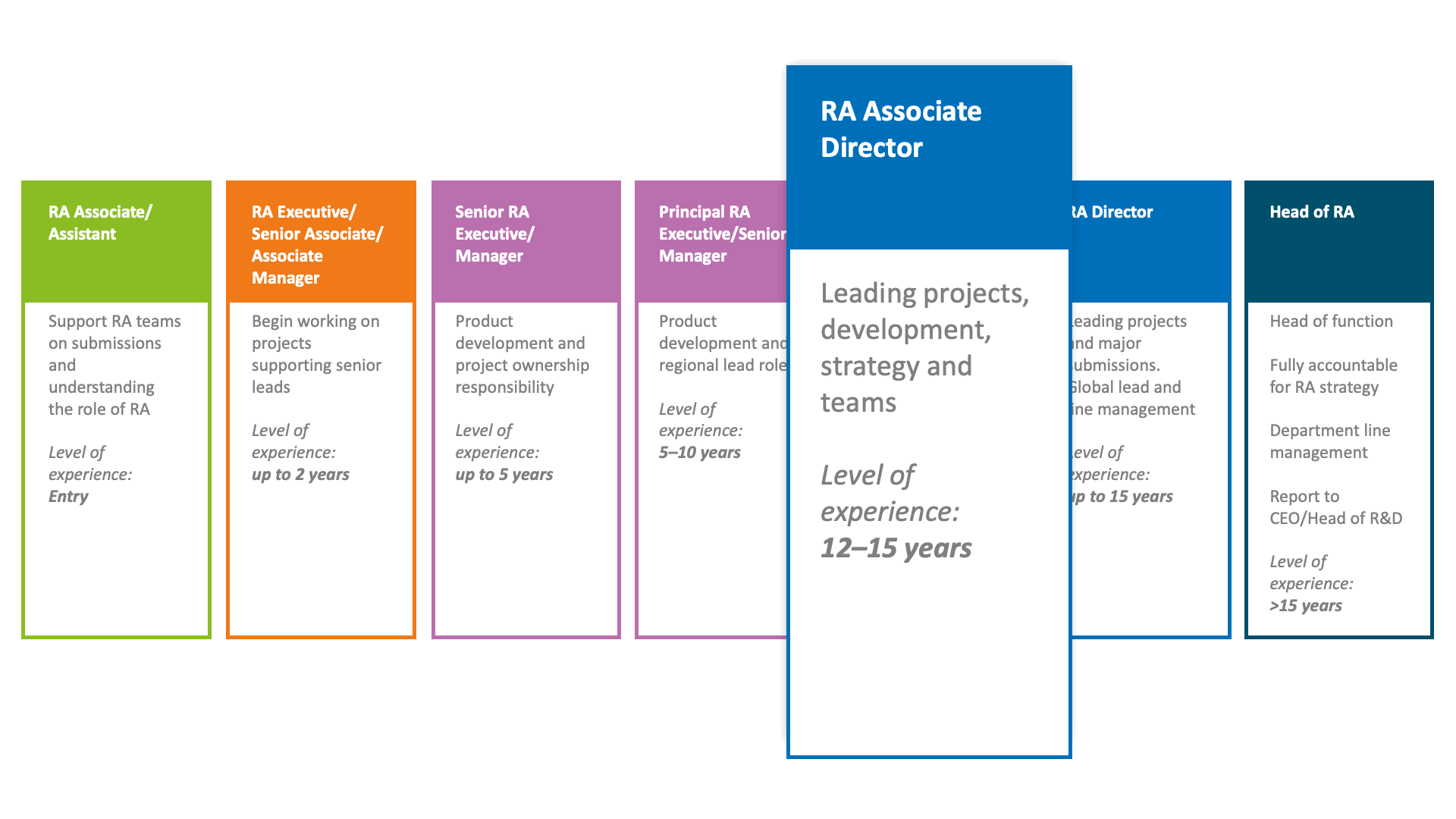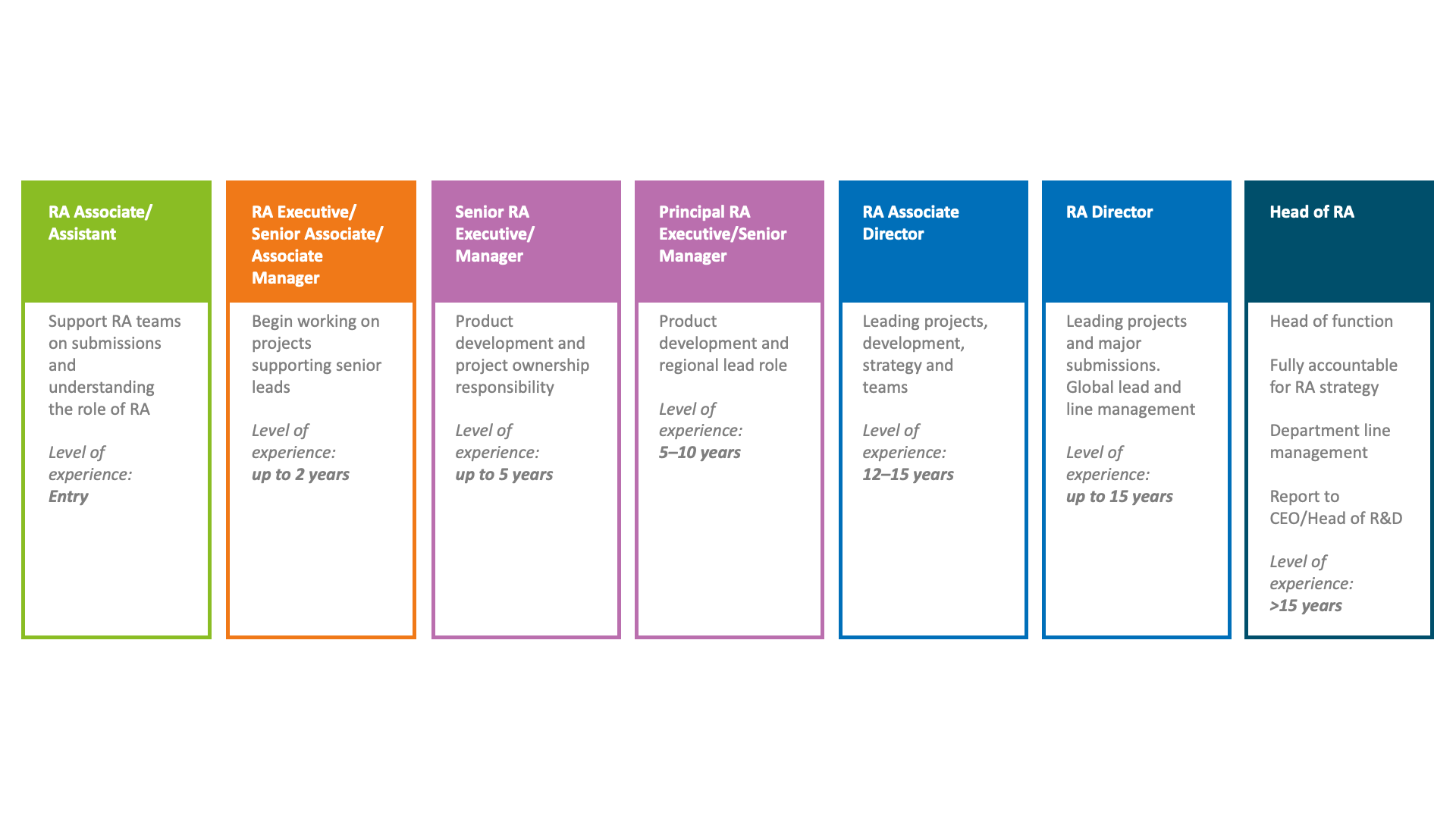
Some examples of the varied tasks that regulatory professionals may undertake as part of their everyday roles:
- Develop regulatory and development strategies for new and existing products
- Analyse data to ensure suitability for regulatory submission
- Put together regulatory submissions (eg, clinical trial applications, licence applications and submissions to change information)
- Write parts of regulatory dossiers
- Represent regulatory affairs on global project development team
- Project manage a development or submission programme
- Manage products post-licensing
- Manage a product in a particular region or territory
- Lead health authority (HA) meetings on behalf of your company (eg, scientific advice meetings, oral explanations for MAA)
- Supporting regulatory inspections from health authorities
- Overseeing overall company regulatory compliance.
The following are examples of different types of role for experienced regulatory professionals.
Geographic roles
These vary from local affiliate roles in a specific country, to regional leads and global leads. Each differs in their day-to-day tasks and may require different expertise.
Local affiliates
- Manage regulatory activities for a country or region (eg, Benelux - Belgium, Netherlands and Luxembourg)
- Can be local employees within subsidiaries/affiliates of the parent organisation or third parties (eg, CRO)
- Most of role is post-approval regulatory work
- Maintain relationships with the health authorities, developing in-depth knowledge of local regulations and requirements
- Interpret the regulatory strategy and communicates to country(ies)
- Manage local variations and renewals, reviews artwork, etc
- Maintain close relationships with local commercial, medical affairs and market access functions.
Regional leads
- Manage regulatory activities for a region (eg, EU)
- Sit within the head office of the parent organisation or in a local office
- Can cover both pre- and post-approval work
- Maintain relationships with regional health authorities (eg, EMA), developing in-depth knowledge of regional regulations and requirements
- Contribute to developing the company’s global regulatory strategy
- Manage regional (centralised/MRP/DCP) variations, renewals and/or core packages for their region
- Maintain close relationships with CMC, regional commercial, medical affairs and market access.
Global leads
- Usually sit within a head office of the parent organisation in either the EU or the US
- Can cover both pre- and post-approval products
- May liaise with health authorities if also taking a regional lead role (eg, US lead)
- Liaise with regional leads and are accountable for developing the global regulatory strategy, including interactions with regulatory authorities
- Communicate global regulatory strategy to regional leads and support execution in regions as necessary
- Maintain close relationships with regional regulatory, global commercial, medical affairs and market access
- Usually part of the core global project team – close relationships with all global colleagues such as clinical, nonclinical, CMC, biostats, project leads/management
- Usually responsible for managing applicable internal governance activities.
Across the lifecycle roles
Generally speaking, pre-approval work revolves around bringing a new product to the market, whereas post-approval work focuses on maintaining compliance of the authorised licence.
Pre-approval – or development regulatory affairs
- Pre-approval work aims to bring a new or updated product through nonclinical and clinical development according to the applicable global and regional regulations
- The goal is to generate data packages pre-authorisation that are ultimately directly or indirectly sufficient to support a licence application referred to as a New Drug Application (NDA) in the US, Japan, Korea, etc, or marketing authorisation application (MAA) in the EU and many other global markets (EU)/other key markets (Japan, China, etc), or medical device certification/clearance/approval in the many global markets
- Generating and updating regulatory strategy as development milestones progress to optimise registration of the target product label
- Providing regulatory considerations to inform country choice for clinical trial sites, advising on regulatory requirements for clinical trials to better assure successful licence applications, submitting CTAs (EU) and Investigational New Drug applications and amendments (US)
- Managing HA interactions, nationally (eg, FDA) and regionally (eg, EMA, CAs/NBs in case of medical devices) which include regulatory scientific advice meetings alone or in parallel with health technology assessment bodies (who deal with pricing and reimbursement of medicines/medical devices)
- Strong understanding and familiarity of the requirements of International Conference for Harmonisation (ICH) and applicable regional guidelines available
- Close relationships with global colleagues including clinical, CMC, biostatistics and global/regional regulatory colleagues, commercial and safety.
Post-approval – or lifecycle management
- Post-approval work aims to further develop and ensure the maintenance on the market of a product that has already received approval for at least one indication.
- The product can be further developed through collection of nonclinical, clinical development or additional data (real-world evidence) across the product lifetime which can lead to updated information on the product via lifecycle management activities in line with the applicable global, regional and local regulations, including (but not limited to):
- Variations (labelling, CMC, etc)
- Design changes and field actions
- Renewal of the licence
- Post-authorisation commitments
- Required safety reports (eg, Periodic Safety Update Reports)
- Annual reports for CMC
- Or aim at further developing a product via extension of the licence (eg, new indication).
- When necessary and for further development purposes: submitting CTAs and INDs or amendments
- Managing HA interactions, nationally and regionally, or locally when applicable, which may range from dealing with queries through to scientific advice
- Strong understanding and familiarity of the requirements of ICH and applicable regional guidelines available
- Close relationships with global colleagues including clinical, CMC, biostatistics and global/regional regulatory colleagues.